In a recent study published in the United States Centers for Disease Control and Prevention (CDC) Emerging Infectious Diseases journal, researchers utilize a whole-genome surveillance (WGS) protocol from the National Taiwan University (NTU) to monitor community transmission and evolution of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in Taiwan.
Study: Emergence and persistent dominance of SARS-CoV-2 Omicron BA.2.3.7 variant, Taiwan. Image Credit: Lightspring / Shutterstock.com
Background
The SARS pandemic hit Taiwan nearly two decades before the coronavirus disease 2019 (COVID-19) pandemic occurred. Based on their prior experience, this small island country quickly contained the few SARS-CoV-2 outbreaks that occurred, with most Taiwanese citizens remaining SARS-CoV-2 naive until 2022.
However, in April 2022, Taiwan recorded 17,000 confirmed COVID-19 cases, most of which were due to the SARS-CoV-2 Alpha variant. At this time, most Taiwanese residents had received COVID-19 vaccines. From northern Taiwan, Omicron infection clusters spread throughout the country, with new case numbers exceeding 50,000 per day.
Taiwan provides a unique opportunity to study how Omicron and its subvariants disseminated throughout the country, despite strict mitigation measures, such as travel restrictions for passengers arriving by air or sea and compulsory quarantine for some people entering the country.
About the study
In the present study, researchers performed WGS on nasal swab samples found to be SARS-CoV-2-positive on reverse transcriptase-polymerase chain reaction (RT-PCR) assay. Since the genomic data was high quality, most sequenced samples showed over 98% coverage of the SARS-CoV-2 target genome.
Subsequently, the researchers assembled all SARS-CoV-2 genome sequences for viral lineages and performed phylogenetic analysis using the Pango-dynamic nomenclature system. This allowed for signature mutations to be closely monitored in all viral samples, including nonsynonymous amino acid (AA) changes in the Omicron samples.
A total of 1,966 genome sequences from the study sample were compared against 881 sequences obtained from the Global Initiative on Sharing Avian Influenza Data (GISAID) to construct a phylogenetic tree. From these data, 1,577 Omicron sequences from the current study were compared against 228 Omicron BA.2.3.7 sequences from the GISAID database.
Study findings
Three AA changes were observed, including L631F in the open reading frame (ORF) 1a, M322I in the nucleocapsid protein, and K97E in spike (S) glycoprotein in the fourth sequencing batch; however, these changes were absent in batches three to five. The genomic sequences containing the signature mutations steadily increased from 62% to 85% between batches four and five.
Notably, Omicron BA.1 or BA.2 subvariants, which contained the three aforementioned AA mutations, constituted batch-three isolates. Thus, BA.2.3.7 likely emerged due to a founder effect and, as a result, was responsible for the dramatic surge in COVID-19 cases in Taiwan from zero to about 100,000 cases/day.
Adequate evidence for the presence and wide-scale transmission of this novel SARS-CoV-2 lineage BA.2.3.7 in Taiwan was also reported. BA.2.3.7, first identified on March 27, 2022, continuously evolved, as reflected in the multiple genomic changes detected since this date. For example, BA.2.3.7 acquired a novel mutation, G1251V, in the S protein from April 2022 onward that later rapidly disseminated across Taiwan.
Genetics & Genomics eBook

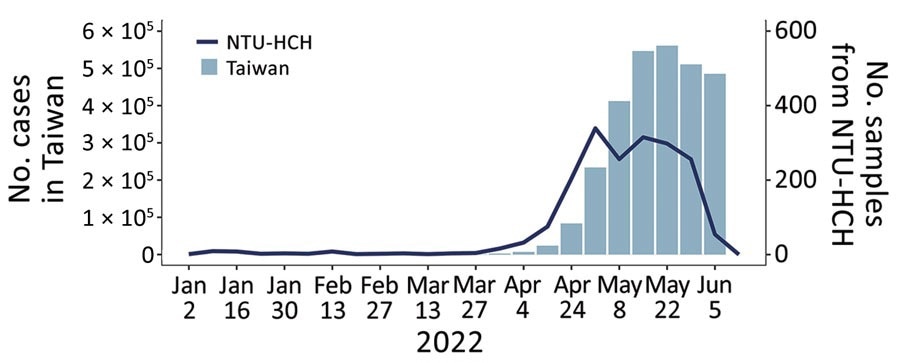
Weekly statistics for confirmed COVID-19 cases in Taiwan and sequenced samples, lineage distribution, and mutation prevalence derived from the NTU-HCH surveillance program, January–June 2022. Graph shows the number of COVID-19 confirmed cases in Taiwan and the sequenced samples from NTU-HCH from January (epidemiologic week 1) to early June (epidemiologic week 23). This figure was constructed using publicly available data of Taiwan Centers for Disease Control (https://nidss.cdc.gov.tw/nndss/disease?id=19CoV). NTU-HCH, National Taiwan University Hospital–Hsinchu Branch.
Conclusions
The current study surveyed one medical center in Taiwan, during which the number of COVID-19 cases was relatively small in this country. However, four other locations in Taiwan also reported Omicron BA.2.3.7 cases, thus suggesting that this novel SARS-CoV-2 Omicron sublineage was likely present across the island nation.
BA.2.3.7 subsequently traveled from Taiwan to other Asia-Pacific countries, such as Japan; however, it did not spread globally or to other countries, such as the United States of America.
To summarize, the current study demonstrated how a COVID-19 outbreak occurred in Taiwan's naive and vaccinated population, despite strict travel restrictions. Researchers believe that genetic drift, which is otherwise known as the founder effect, or the ability of Omicron BA.2.3.7 to evade vaccine-induced immunity, contributed to the rapid emergence of this subvariant in Taiwan.
As Asian countries are easing restrictions for international travelers and shifting from zero tolerance to less stringent COVID-19 policies, it remains necessary to continue SARS-CoV-2 surveillance using WGS methodologies. In addition, early detection of new SARS-CoV-2 variants in endemic countries like Taiwan could help minimize future disruptions due to COVID-19 worldwide.
- Shao, P., Tu, H., Gong, Y., et al. (2023). Emergence and persistent dominance of SARS-CoV-2 Omicron BA.2.3.7 variant, Taiwan. Emerging Infectious Diseases. doi10.3201/eid2904.221497
Posted in: Genomics | Medical Science News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Assay, Avian Influenza, Coronavirus, Coronavirus Disease COVID-19, covid-19, Evolution, Genetic, Genome, Genomic, Glycoprotein, immunity, Infectious Diseases, Influenza, Mutation, Omicron, Pandemic, Polymerase, Polymerase Chain Reaction, Protein, Respiratory, Reverse Transcriptase, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Vaccine

Written by
Neha Mathur
Neha is a digital marketing professional based in Gurugram, India. She has a Master’s degree from the University of Rajasthan with a specialization in Biotechnology in 2008. She has experience in pre-clinical research as part of her research project in The Department of Toxicology at the prestigious Central Drug Research Institute (CDRI), Lucknow, India. She also holds a certification in C++ programming.
Source: Read Full Article


